肺癌是世界上最常见的恶性肿瘤之一,已成为我国城市人口恶性肿瘤死亡原因的第1位。非小细胞型肺癌(NSCLC)包括鳞癌、腺癌、大细胞癌,与小细胞癌相比其癌细胞生长分裂较慢,扩散转移相对较晚。非小细胞肺癌约占所有肺癌的80%,约75%的患者发现时已处于中晚期。
表皮生长因子受体(Epidermal growth factor receptor,EGFR)是一种跨膜蛋白,由细胞外的配体结合区、疏水跨膜结构域和细胞内的激酶区3部分组成,其胞内结构包含1个酪氨酸激酶域和具有多个自身磷酸化位点的羧基末端,属于受体酪氨酸激酶家族。EGFR对于控制上皮细胞的生长和存活至关重要,通常用于治疗非小细胞肺癌(NSCLC)等上皮恶性肿瘤。
当前,包括小分子酪氨酸激酶抑制剂(TKIs)和靶向EGFR的
单克隆抗体都集中于抑制EGFR激酶活性或诱导抗体和补体介导的细胞毒性作用。其中靶向EGFR的抗体主要用于治疗晚期结直肠癌和头颈部肿瘤,但其临床治疗效果不佳,因此不适用于治疗NSCLC。迄今为止已经开发了三代EGFR 酪氨酸激酶抑制剂(TKIs) 可逆地(第一代)或不可逆地(第二代和第三代)抑制EGFR酪氨酸激酶活性,并已广泛用于NSCLC治疗。
吉非替尼和厄洛替尼是针对野生型EGFR设计的第一代EGFR TKIs,但对活性EGFR突变显示出强大的的选择性抑制作用。
二代EGFR TKIs(如阿法替尼、达可替尼)被设计用于克服一代EGFR TKIs的获得性T790M耐药性,但由于毒性不可接受失败了。
新开发的第三代EGFR TKIs(如奥希替尼和诺司替尼)与EGFR ATP结合位点上的半胱氨酸-797残基不可逆结合,相对于WT-EGFR而言,对携带激活的突变或T790M耐药突变的EGFR形式显示出优先的活性。
大量研究表明,EGFR空间分布和稳定性也是调节肺癌进展的关键决定因素。即使在突变的EGFR驱动的肺腺癌中,EGFR降解失调也进一步加速了肿瘤的发生和发展。表皮生长因子受体的空间失调增加了质膜受体的可用性,并诱导持续的信号输出。甾醇-C4-甲基氧化酶样和NAD(P)H类固醇脱氢酶样蛋白的消耗,这两种蛋白都参与了甾醇生物合成途径,抑制了EGFR的再循环,并使A431异种移植物对西妥昔单抗治疗敏感。高尔基体膜蛋白1与EGFR相互作用,促进EGFR循环至膜,导致EGFR激活时间延长和肝细胞癌进展。这些发现都强调了促进EGFR降解是靶向EGFR相关癌症的一种替代策略。

近日一项研究确定了TRIB3表达的升高以及EGFR的稳定性、再循环、信号活性与NSCLC进展的增加有关,揭示了通过加速EGFR降解来干扰TRIB3-EGFR相互作用治疗NSCLC的潜在作用。这项研究结果由中国医学科学院药物研究所胡卓伟研究员和花芳研究员带领的团队发现,并发表在Nature杂志上。
TRIB3作为压力传感器可以响应各种压力源,通过与信号蛋白与功能蛋白的相互作用来参与慢性炎症、代谢和恶性疾病。研究证实,TRIB3通过与自噬受体p62相互作用,削弱了自噬和蛋白酶体的降解功能,从而促进了多种癌症的发生和发展。TRIB3的缺失会导致各类癌症中多种肿瘤促进因子(包括EGFR)的表达显著下降。
TRICL3表达与NSCLC中的EGFR呈正相关
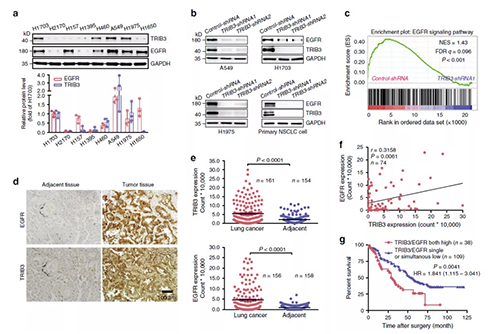
在指定的NSCLC细胞系中对TRIB3和EGFR表达的免疫印迹分析(来源:Nature)
为了确定TRIB3和EGFR水平在肺癌中的关系,研究人员检测了这两种蛋白质在几种人类肺癌细胞系中的表达。在大多数人NSCLC细胞系中,TRIB3的高表达与EGFR的表达升高相关(a)。TRIB3耗竭不仅降低了这些细胞系和原代NSCLC细胞中的EGFR表达(b),而且抑制了A549细胞中的EGFR反应基因(c)。
研究人员使用在线kmplot工具查询了TCGA数据库对1416名NSCLC患者进行评估,并确定高TRIB3 mRNA水平仅与肺腺癌的不良生存相关(a),但不包括肺鳞癌(b)。但是,发现高TRIB3蛋白与肺腺癌(c,d)和鳞癌中的不良存活率呈正相关。与TRIB3蛋白表达一致,在人NSCLC组织样品中观察到的EGFR蛋白水平高于相邻的非肿瘤组织样品(d,e)。在NSCLC组织中,TRIB3与EGFR蛋白水平之间存在正相关(f)。值得注意的是,在同时表达EGFR和TRIB3患者的存活率显著低于单一表达表达EGFR或TRIB3和同时低表达的患者(g)。
TRIB3促进PKCα介导的EGFR Thr654磷酸化

TRIB3与EGFR相互作用以促进PKCα介导的EGFR磷酸化(来源:Nature)
TRIB3通过蛋白相互作用诱导多种细胞功能,研究人员进行了高通量蛋白质阵列筛选并且鉴定PKCα是TRIB3的结合伴侣(a)。
共免疫沉淀(CO-IP)分析表明,TRIB3、EGFR和PKCα被每种抗体共沉淀(b)。
另外,在EGF刺激后TRIB3、EGFR和PKCα共定位在细胞质中(c)。
据了解,EGFR中的Thr654是PKCα的主要磷酸化位点,在EGF处理下,对照A549细胞中大量EGFR与PKCα共定位(d左);
在TRIB3缺失的细胞中,共定位比对照细胞少(12±2%),并伴有EGFR和PKCα总量减少(d右)。
即使当PKCα异位表达时,TRIB3消耗也会减少EGF诱导的EGFR T654磷酸化(e)。
为了绘制与EGFR和PKCα相互作用的TRIB3的相互作用区域,构建了带有HA标签的TRIB3的缺失突变体并进行了CO-IP测定,鉴定出TRIB3激酶死亡区域的C末端与EGFR相互作用(f)。
TRIB3的C末端尾部负责TRIB3和PKCα之间的结合(g)。
此外,鉴定出EGFR的细胞内近膜区域与TRIB3的相互作用(h,i)。
恢复TRIB3的表达而不恢复KDC缺失突变体(M5)可以逆转TRIB3耗竭对EGFR循环的抑制作用(j)。
这些数据均表明TRIB3和EGFR之间的相互作用对于EGFR循环至关重要。
EGFR靶向降解抑制肺癌的发展
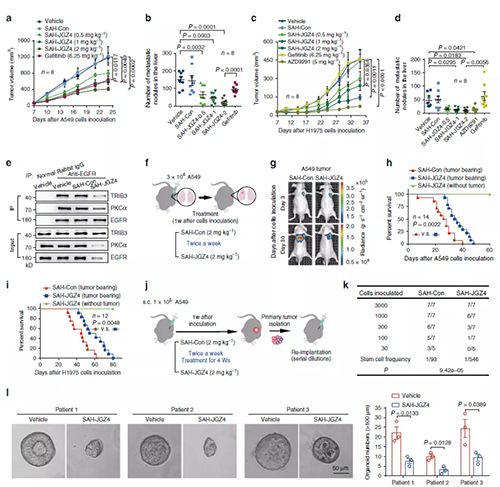
靶向EGFR稳定性抑制肺癌的发生和发展(来源:Nature)
研究人员发现SAH-JGZ4可以促进EGFR降解并抑制EGFR信号传导活性,因此使用体外和体内模型评估了其抗肿瘤作用。SAH-JGZ4不仅可以抵抗EGF诱导的核心多能性因子的表达(g),而且还可以抑制A549细胞中的肿瘤增殖、侵袭和内在肿瘤形成(h-j)。SAH-JGZ4处理以剂量依赖性方式抑制具有A549细胞的皮下异种移植模型中的肿瘤生长,并且与吉非替尼相比,每周两次给药2 mg kg -1的剂量具有更好的抗肿瘤作用(a)。SAH-JGZ4诱导了肝转移的剂量依赖性降低,这优于吉非替尼诱导的降低(b)。值得注意的是,SAH-JGZ4还抑制了接种
NCI-H1975细胞的小鼠的肿瘤生长和转移,NCI-H1975细胞是具有T790M突变的吉非替尼耐药肺癌细胞(c,d);SAH-JGZ4的抗肿瘤功效优于吉非替尼,且与AZD9291相当(图 c,d)。从机制上讲,SAH-JGZ4干扰了EGFR–TRIB3和EGFR–PKCα的体内相互作用,并抑制了接种肿瘤组织中EGFR和PKCα的表达(图e)。此外,用SAH-JGZ4处理的接种NCI-H1975小鼠的肿瘤组织样品中STAT3 / 5和EGFR的磷酸化以及总EGFR、PKCα、TRIB3和核心多能因子的表达降低了(d,E)。使用肺原位移植模型,我们发现SAH-JGZ4从接种侧向相反侧减弱了A549细胞的转移(图f,g)。SAH-JGZ4将肿瘤起始细胞(TIC)的频率降低了六倍(图j,k),表明靶向EGFR的稳定性是抑制肺癌干细胞生长的潜在策略。
结果与讨论
这项研究表明升高的TRIB3通过增强EGFR的循环利用和稳定性来参与NSCLC的发病和进展。TRIB3–PKCα–WWP1通过在K689上诱导EGFR的K63连接泛素化作用,对EGFR的回收利用和稳定性形成了积极的调节轴。此外,研究人员通过诱导K63连接的K689 EGFR泛素化来显示有关WWP1的肿瘤促进作用的分子细节,随后促进EGFR循环并维持EGFR稳定性。
同时在此项研究中还开发了SAH-JGZ4,其可以干扰TRIB3-EGFR的相互作用,并通过抑制EGFR再循环并随后诱导EGFR降解而产生抗癌作用。此外,SAH-JGZ4处理还通过促进c-Met降解并持续抑制STAT3 / 5信号转导而对补偿途径表现出抑制作用。EGFR和KRAS突变是NSCLC中的两个主要驱动因素,与EGFR靶向疗法相反,临床上没有有效的针对突变KRAS蛋白的抑制剂,而此次研究发现SAH-JGZ4治疗KRAS突变的A549细胞会降低KRAS活性,并抑制肿瘤的发生和发展,这表明促进EGFR降解具有靶向“不可治疗的” KRAS突变的治疗潜力。SAH-JGZ4的这些特征表明,它是针对EGFR靶向治疗(尤其是克服TKI耐药性)的有效替代药物,研究人员表示SAH-JGZ4与KRAS抑制剂联合使用的治疗效果也会在未来的研究中继续进行评估。
总而言之,研究表明TRIB3、EGFR和PKCα的协同表达和作用建立了TRIB3-PKCα-WWP1调控轴,通过增强EGFR的再循环、稳定性和信号传导来促进NSCLC的发展。此外,靶向TRIB3-EGFR相互作用以促进EGFR降解是治疗EGFR相关NSCLC病例的潜在治疗选择,美迪西作为新药研发CRO也会持续关注此项研究进展,希望能为肺癌新药的研发助力。
关于美迪西
美迪西(股票代码:688202)成立于2004年,总部位于上海,致力于为全球制药企业、研究机构及科研工作者提供全方位的临床前新药研究服务。美迪西的一站式综合服务以强有力的项目管理和更高效、高性价比的研发服务助力客户加速新药研发进程,服务涵盖医药临床前新药研究的全过程,包括药物发现、药学研究及临床前研究。美迪西与国内外优质客户共同成长,为全球超过700家客户提供新药研发服务,美迪西将继续立足全球视野,聚力中国创新,为人类健康贡献力量!
联系我们
Email: marketing@medicilon.com.cn
电话: +86 (21) 5859-1500(总机)




















