



一、蛋白质表达与纯化实验原理
(一)利用大肠杆菌表达进行原核表达
基因工程的最终目的是在一个合适的系统中,使外源基因高效表达,从而生产有价值的蛋白质产品。
1.原核生物基因表达的特点
(1)原核生物(如大肠杆菌)只有一种RNA聚合酶,识别原核的启动子,能够催化所有RNA合成。
(2)原核生物的基因表达是以操纵子为单位。操纵子是数个相关的结构基因及其调控区的结合,是一个基因表达的协同单位。
(3)由于原核生物无核膜,所以转录与翻译是耦联的,二者也是连续进行的。原核生物染色体DNA是裸露的环型DNA,转录成mRNA后,可直接在胞浆中与核糖体结合翻译成蛋白质。每个核糖体可独立完成一条链的合成,多核糖体可同时在一条mRNA合成多条肽链,大大提高了翻译效率。
(4)原核基因不含内含子(intron),没有转录后加工过程。
(5)原核生物基因表达的控制主要是在转录水平。对RNA合成的调控有两种方式:启始控制(启动子控制)和终止控制(衰减子控制)。
(6)在大肠杆菌mRNA的核糖体结合位点上,有一个转译启始密码子及同16S核糖体RNA 3′末端碱基互补的序列,即SD序列,真核基因则无此序列。
2.外源基因在原核中表达的关键
(1)表达载体将外源基因导入宿主菌,指导宿主菌的酶系统合成外源蛋白;
(2)外源基因不能带有内含子;
(3)利用原核细胞的强启动子和SD序列等控制外源基因的表达;
(4)外援基因与表达载体连接后,必须形成正确的开放阅读框架;
(5)利用宿主菌的调控系统,调节外源基因的表达,防止表达的外源基因产物对宿主菌的毒害。
3.外源基因在原核细胞中表达的重要调控元件
(1)启动子
-35区:位于转录启始位点上游35bp处,一般由10bp组成,与RNA聚合酶δ亚基结合;
-10区(TATA box):位于转录启始位点上游5-10bp处,一般由6-8bp,富含A T,与RNA聚合酶的核心酶结合。
原核表达载体所用的启动子必须是原核启动子,通常所用的可调控的强启动子有:lac(乳糖启动子)、trp(色氨酸启动子),λPL(λ噬菌体左向启动子),Tac(乳糖和色氨酸的杂和启动子)等。
(2)SD顺序
mRNA在细菌中转译率严格依赖于SD序列以及SD序列与启始密码子AUG之间的距离,例如,当lac启动子的SD序列距AUG为7个核苷酸时,IL-2表达最高,为2581单位,而间隔8个核苷酸时,表达水平降到不足5个单位。另外某些蛋白质与SD序列结合也回影响蛋白质的翻译。
(3)终止子(terminator)
4.原核表达载体的类型
(1)非融合型
不与细菌的任何蛋白或多肽融合在一起的表达蛋白。
优点:非常接近于生物体内天然蛋白质;
缺点:容易被细菌蛋白酶破坏,未知蛋白难于纯化。
(2)融合型
蛋白质的一端由原核DNA序列或其它序列编码,另一端由真核DNA的完整序列编码。
优点:应该避免细菌蛋白酶的破坏,由于融合部分的关系,常易于表达产物的分离纯化。
缺点:由于细菌的一段蛋白或多肽的存在,有时会影响其结构,需去掉。
5.用于原核细胞表达的外源基因
由于原核细胞缺乏真核细胞转录后的加工系统,mRNA的内含子不能切除,成熟的mRNA不能形成,同时原核细胞也缺乏真核细胞翻译后的加工系统。只能用cDNA而不能用基因组DNA,或体外合成基因,PCR扩增基因等。
(二)利用HIC树脂纯化ZsGreen蛋白
甲基化疏水反应层析(HIC)树脂是利用疏水基团进行分离蛋白的一种简单而有效的方法。结合缓冲液中的Cl-排斥带负电荷的ZsGreen分子的β-外壳,这种排斥使分子内部外翻,露出疏水基团。露出的疏水基团牢牢与HIC树脂的非极性甲基基团结合;中性盐洗脱使ZsGreen保持为疏水状态;但是可以洗脱未结合或者是结合弱的蛋白;低盐缓冲液使疏水基团回到ZsGreen分子的原来位置,从而使ZsGreen分子HIC树脂中释放。
(三)SDS-PAGE分析
聚丙烯酰胺凝胶电泳是网状结构,具有分子筛效应,它有两种形式,一种是非变性聚丙烯酰胺凝胶,蛋白质在电泳中保持完整的状态,蛋白在其中依三种因素分开:蛋白大小,形状和电荷。而SDS-PAGE仅根据蛋白分子量亚基的不同而分离蛋白。这个技术首先是1967年由shapiro建立,他们发现在样品介质和丙烯酰胺凝胶中加入离子去污剂和强还原剂后,蛋白质亚基的电泳迁移率主要取决于亚基分子量的大小,电荷因素可以忽视。
SDS是阴离子去污剂,作为变性剂和助溶试剂,它能断裂分子内和分子间的氢键,使分子去折叠,破坏蛋白分子的二、三级结构。而强还原剂如巯基乙醇,二硫苏糖醇能使绊胱氨酸残基间的二硫键断裂。在样品和凝胶中加入还原剂和SDS后,分子被解聚成多肽链,解聚后的氨基酸侧链和SDS结合成蛋白——SDS胶束,所带的负电荷大大超过了蛋白原有的蛋白量,这样就消除了不同分子间的电荷差异和结构差异。SDS-PAGE一般采用的是不连续缓冲系统,于连续缓冲系统相比,能够有较高的分辨率。
浓缩胶的作用是有堆积作用,凝胶浓度较小,孔径较大,把较稀的样品加在浓缩胶上,经过大孔径凝胶的迁移作用而被浓缩至一个狭窄的区带。当样品液和浓缩胶选Tris-HCl缓冲液,电极液选Tris-甘氨酸。电泳开始后,HCl解离成Cl-,甘氨酸解离出少量的甘氨酸根离子。蛋白质带负电荷,因此一起向正极移动,其中Cl-最快,甘氨酸根离子最慢,蛋白居中。电泳开始时Cl-泳动率最大,超过蛋白,因此在后面形成低电导区,而电场强度与低电导区成反比,因而产生较高的电场强度,使蛋白和甘氨酸根离子迅速移动,形成以稳定的界面,使蛋白聚集在移动界面附近,浓缩成一中间层。
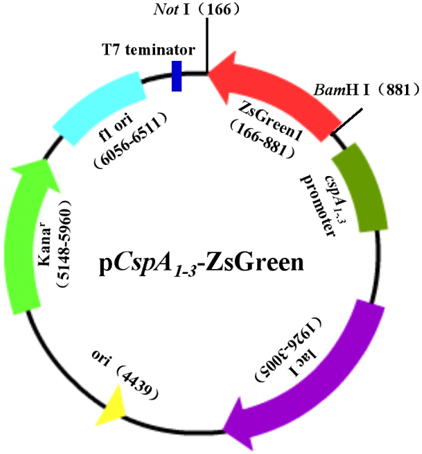
本实验中,我们首先提取大肠杆菌(E.coli)基因组,PCR扩增冷激蛋白CspA的启动子、上游调控及下游5′-UTR区序列,得到cspA1、cspA2和cspA3,在两端引入酶切位点;用限制性内切酶切割质粒pZsGreen1-1,得到绿色荧光蛋白ZsGreen报告基因;以pET-28a(+)为骨架,酶切、连接,构建含冷激启动子、报告基因为ZsGreen的表达载体pCspA1-ZsGreen、pCspA2-ZsGreen、pCspA3-ZsGreen(图1)及对照常温表达载体pT7-ZsGreen(图5-1)。
二、实验仪器及药品 (1)STE的配制(表3-1)
表3-1STE(100mL)配方
| 试剂名称 | 试剂等级 | 用量 | 最终浓度 |
5mol/L NaCl | 分析纯 | 2mL | 0.1mol/L |
1mol/L Tris-HCl(pH8.0) | 分析纯 | 1mL | 10mmol/L |
0.5mol/L EDTA(pH8.0) | 分析纯 | 200μL | 1mmol/L |
dH2O | 不需灭菌 | up to 100mL | — |
在60mL dH2O中加入5mol/L NaCl 2mL、1mol/L Tris-HCl(pH8.0) 1mL和0.5mol/L EDTA(pH8.0)200μL,混匀,加dH2O定容至100mL,1.034×105Pa高压蒸汽灭菌15min,备用。配制好的STE含有0.1mol/L NaCl,10mmol/L Tris-HCl和1mmol/L EDTA。
(2)溶液I的配制(表3-2)
表3-2溶液I(100mL)配方
| 试剂名称 | 试剂等级 | 用量 | 最终浓度 |
葡萄糖 | 分析纯 | 0.9g | 50mmol/L |
1mol/L Tris-HCl(pH8.0) | 分析纯 | 2.5mL | 25mmol/L |
0.5mol/L EDTA(pH8.0) | 分析纯 | 2mL | 10mmol/L |
dH2O | 不需灭菌 | up to 100mL | — |
60mL dH2O中加入0.9g葡萄糖、1mol/L Tris-HCl(pH8.0) 2.5mL和0.5mol/L EDTA(pH8.0) 2mL,混匀,加dH2O定容至100mL,1.034×105Pa高压蒸汽灭菌15min,4℃贮存。配制好的溶液I含有50mmol/L葡萄糖,25mmol/L Tris-HCl和10mmol/L EDTA。
(3)溶液II的配制(表3-3)
溶液II中含有0.4mol/L NaOH和1% SDS,该试剂需要新鲜配制,配制后将NaOH和SDS按1:1的比例混合。
表3-3溶液II(1mL)配方
| 试剂名称 | 试剂等级 | 用量 | 最终浓度 |
5mol/L NaOH | 分析纯 | 40μL | 0.4mol/L |
dH2O | 不需灭菌 | up to 500μL | — |
2%SDS | 分析纯 | 500μL | 1% |
dH2O | 不需灭菌 | up to 500μL | — |
(4)溶液III的配制(表3-4)
表3-4溶液III的配制(100mL)
| 药品名称 | 用量 |
5mol/L KAc | 60mL |
冰醋酸 | 11.5mL |
dH2O | 28.5mL |
配制好的溶液III含3mol/L KAc、5mol/L冰醋酸(pH4.8)。
三、质粒抽提实验方法
质粒提取过程如下:
(1)将琼脂培养板上的阳性单菌落,移至3mL LB液体培养基中(含Amp或Kana),或将大肠杆菌的甘油菌按1:30的比例接到LB液体培养基中,37℃剧烈摇荡培养过夜;
(2)取500μL菌体,与40%甘油等体积混合,-70℃保存,剩余的菌体用于质粒的提取;
(3)取出1.5-2mL菌液移至Eppendorf管中,12000rpm,4℃离心30s,弃上清,用1mL STE悬浮菌体沉淀,洗涤菌体,再离心回收菌体;
(4)再重复用STE漂洗菌体,离心后,去尽上清液;
(5)将细菌沉淀悬浮于100μL冰预冷的溶液I中,强烈振荡混匀;
(6)加入200μL新配制的溶液II,轻轻颠倒离心管5次以混合内容物,不要强烈振荡,放置冰上3min左右(根据不同菌株,可适当缩短);
(7)加入150μL冰预冷的溶液III,轻轻颠倒5次,混匀,置于冰上5min;
(8)15000rpm,4℃离心5min,将上清转移到另一个离心管中;
(9)向上清中加入等体积的酚:氯仿(1:1),混匀,15000rpm离心5min,将上清转移到另一个离心管中;
(10)加入2倍体积冰预冷的无水乙醇,室温放置2min,沉淀双链DNA;
(11)15000rpm,4℃离心5min;
(12)倒掉上清,使液体尽量流尽,空气中干燥沉淀;
(13)用20μL含有RNaseA的TE缓冲液溶解核酸沉淀,瞬时离心,混匀,贮存于-20℃冰箱中备用。
四、实验结果
(一)未纯化SDS-PAGE分析
将含有pCspA1-ZsGreen、pCspA2-ZsGreen、pCspA3-ZsGreen的大肠杆菌(E.coli),在16℃表达;含有pT7-ZsGreen的大肠杆菌(E.coli)在37℃下表达至OD600=0.6时,加入IPTG诱导表达,不加IPTG诱导的作为对照。诱导表达5h后收集菌体,进行SDS-PAGE分析(图5-2)。
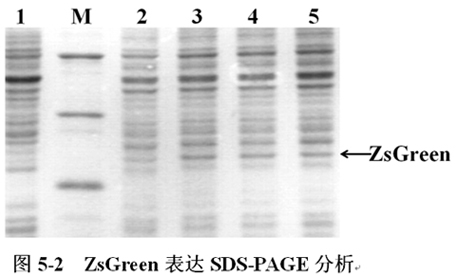
图5-2中,M为低分子量蛋白Maker。1-5分别为pT7-ZsGreen(IPTG-,37℃)、pT7-ZsGreen(IPTG+,37℃)、pCspA1-ZsGreen(16℃)、pCspA2-ZsGreen(16℃)、pCspA3-ZsGreen(16℃)表达ZsGreen。箭头所示为ZsGreen,其大小约为27KD。SDS-PAGE结果显示:三种低温诱导表达载体pCspA1-ZsGreen、pCspA2-ZsGreen、pCspA3-ZsGreen在16℃下表达ZsGreen正常,可用作下一步对其翻译效率的分析。
(二)纯化后SDS-PAGE分析
将含有低温诱导表达载体pCspA1-ZsGreen、pCspA2-ZsGreen、pCspA3-ZsGreen的大肠杆菌(E.coli),在16℃下表达,以1h为时间间隔收集菌体,利用HIC树脂纯化ZsGreen蛋白,进行SDS-PAGE分析(图5-3)。
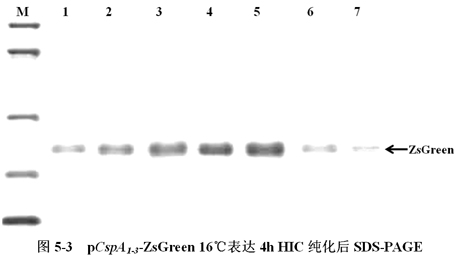
图5-3中,M为低分子量蛋白Maker。A1-7、B8-14、C15-21分别为pCspA1-ZsGreen、pCspA2-ZsGreen、pCspA3-ZsGreen 16℃表达4h HIC纯化后SDS-PAGE。箭头所示为ZsGreen,其大小约为27KD
 相关新闻
相关新闻川沙总部
地址: 上海市浦东新区川大路585号
邮编: 201299
电话: +86 (21) 5859-1500(总机)
传真: +86 (21) 5859-6369
海外:
Email: marketing@medicilon.com
Tel: +1 (617) 888-9294(U.S.)
Tel: 0044 7790 816 954 (Europe)
Tel: +82 70-8269-5849 (Korea)
Tel: +81 80-4421-6898 (Japan)















